BECKWITH-WIEDEMANN SENDROMU
Beckwith-Wiedemann Sendromu (BWS)
bir aşırı büyüme sendromudur. Aşırı büyümeye
neden olan sendromlar arasında en önde gelen BWS, ayı zamanda kanser
oluşturmaya eğilimli durumlar arasında en sık rastlanan hastalıktır.
Genel Özellikler
Görülme sıklığı yaklaşık 10.000’de 1 ila 14.000’de 1’dir,
ancak hafif belirti veren hastalarda BWS tanısının konmadığı düşünülecek
olursa görülme sıklığının daha da fazla olacağı anlaşılır.
Cinsiyet dağılımı eşittir.
İn vitro
fertilizasyon (seçilen sağlam yumurta hücrelerini laboratuvar ortamında
sperm ile buluşturmak) ve intrasitoplazmik sperm injeksiyonu (tek
bir sperm hücresinin doğrudan bir yumurtanın sitoplazmasına enjekte edildiği
bir in vitro fertilizasyon prosedürüdür ve annenin uterusuna nakledilecek
uygun embriyoları elde etmek için kullanılır) gibi çocuk edinme
yöntemlerinin 11. kromozomdaki büyüme üzerinde etkili genleri etkileyerek
BWS riskini on kat arttırdığı ortaya konmuştur.
Nedeni
BWS 11. kromozomda, büyümenin sağlıklı bir şekilde gelişmesi üzerinde etkili
genlerin toplandığı kritik zonda görülen değişik anormallikler sonucu oluşur.
Klinik Görünüm
BW sendromu bulunan hastalar bu sendroma özgü belirtilerin
tümünü, bir kısmını, ya da bir ikisini gösterdiklerinden ve bu klinik
belirtilerin derecesi farklı olduğundan klinik tablo hastadan hastaya
oldukça değişkenlik gösterir. BWS çoğu kez klinik bulgulardan hareketle
yapılan genetik testler sayesinde tanınır. Hatta birçok klinik belirti göze
çarpacak seviyede olmadığından bazen klinik görünüm ile genetik test
sonuçları çelişebilir.
Sendroma yakalanan çocuk normalden daha iridir (makrosomi).
Hastaların yarısından fazlasında doğum tartısı 97 persantilin de üzerindedir.
Normalde doğum sonrası belli olan bu durum bazen daha sonra kendini belli
eder. İri yapı tüm çocukluk dönemince devam eder. Akranlarına göre daha iri
olan bu çocuklarda büyüme yaklaşık 8 yaşına doğru yavaşlamaya başlar.
Erişkin yaşa geldiklerinde artık bu hastalar normal boyuttadırlar.
BWS’lu çocukta asimetrik aşırı büyüme görülür: bu aşırı
büyüme vücudun aynı yarımını kapsadığı gibi (aynı tarafın kol ve/veya bacağı;
hemihipertrofi), çapraz şekilde de olabilir (bir tarafın kolu ve diğer
tarafın bacağı). Asimetrik büyüme yüzde de görülebilir.
Bazı çocuklarda omfalosel (karın
içi organların bir kese içerisinde karnın dışında olmasına yol açan doğumsal
karın duvarı eksikliği), göbek fıtığı, ya da rektus diastazı (diastasis
recti, karın duvarındaki rektus abdominis kaslarının arasının açılmasıyla
orta hatta oluşan karın duvarı zayıflığı) görülebilir.
Bazı çocuklarda doğum sonrası fark edilen, nefes almayı,
yutmayı ve konuşmayı güçleştiren makroglossia (iri dil) bulunur. İri dil alt
çenenin daha belirgin oluşmasına yol açabilir (prognati).
Yeni doğan döneminde saptanan kan şekerinde düşüklük (hipoglisemi)
daha sonraları pankreastaki büyümeye ve artan gereksinime bağlı olarak
insülinin aşırı salgılanması sonucu hiperinsülinemiye yol açabilir.
Hipoglisemiye bağlı belirtiler çoğunlukla fazla olmasa da yeterince tedavi
edilmediğinde zamanla nörolojik komplikasyonlara yol açar.
Karaciğer, dalak, pankreas, böbrekler ve adrenal bezler
gibi batın içi organlarda büyüme (organomegali) görülür.
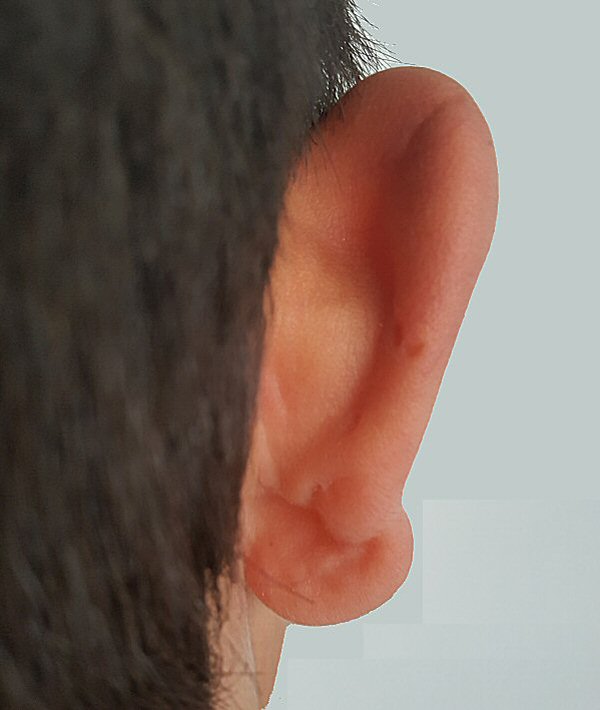
Kulak kepçesinde gamze, minik çukur ve yarık görülmesi
BWS’nun diğer bulgularındandır.
Göz çukuru göreceli olarak daha küçük kaldığından (intraorbital
hipoplazi) hastanın gözleri dışarı fırlak görünümdedir. Mozaizm gösteren
bazı hastalarda her iki gözün renkleri farklı olabilir.
Bazı çocuklar göz kapaklarında ya da alında açık kırmızı
renkte ben tarzı lekelerle doğar. Ufak kan damarlarının toplanmasıyla oluşan
bu benler giderek kaybolur.
Alt ve üst çene kapandığında dişler birbirine uygun bir
şekilde kavuşmaz (maloklüzyon).
Aynı şekilde böbrek anomalileri de görülebilir. Medüller
displazi (böbrek iç dokusunun yeterince gelişmemesi), nefrokalsinoz (böbrekte
kireç çökeltilerinin oluşması), medüller sünger böbrek (kronik böbrek
yetmezliğine yol açan
toplayıcı kanallarda kistik genişlemenin görüldüğü doğumsal bir hastalık)
veya nefromegali (iri böbrek) BWS’nda görülen başlıca
böbrek anomalilerindendir.
BWS’lu hastalarda sinir sisteminin gelişmesi hastalıktan
etkilenmez. Uzun süreli, tedavi görmeyen hipoglisemi, çok erken doğum ve 11.
kromozomda ikileşme (duplikasyon) sinir sisteminin kötüleşmesine neden olan
unsurlardır.
BW sendromlu çocuklar bazı embriyoner tümörlerin gelişmesi
açısından risk altındadırlar. Olguların yaklaşık % 7 ila % 9’u için söz
konusu olan bu oran hemihipertrofinin görüldüğü BWS olgularında çok daha
yüksektir (olguların yaklaşık % 24 ila % 27).
Embriyoner tümörler arasında en sık görülen böbreğin Wilms
tümörüdür; karaciğerin hepatoblastomu bunu izler. Ayrıca nöroblastom (sinir
hücresi tümörü), rabdomiyosarkom (yumuşak doku tümörü) ve adrenal karsinom
(adrenal bezi tümörü) da görülebilir. Bu tümörler hemen her zaman çocukluk
döneminde ortaya çıkar. Yaşamın ilk iki yılı bu embriyoner tümörlerin
gelişmesi açısından en riskli dönemdir.
Hipoglisemi, kardiyomiyopati, makroglossia ve embriyoner
tümörler tedavi edilmedikleri takdirde yaşamı tehdit eder duruma gelirler.
Erken doğuma bağlı komplikasyonları da bunlara eklemek gerekir.
Erişkin yaşa ulaşan BWS’lu hastalarda klinik belirtiler
giderek silikleşir. Erişkinlerin çoğu normal bir görünüme sahiptirler ve
sendroma ait önemli bir sorunla karşılaşmazlar. Yaşam süresi normaldir.
Tanı
BWS hastalarında
moleküler genetik test ile tanı konur.
BWS tanısını
koymak bazen kolay olmaz, çünkü hastaların bir kısmında mozaizm mevcuttur.
Bu nedenle genetik testin değişik dokular üzerinde yapılması tanı koymaya
yardımcı olur.
Tedavi
Merkezi sinir
sistemi komplikasyonlarını önlemek açısından erkenden, daha süt çocukluğu
döneminde hipoglisemi kontrol altına alınmalıdır.
Omfalosel mevcutsa
cerrahi olarak karın duvarı onarımı yapılır.
İri dil nedeniyle
beslenme sorunu yaşayan ufak çocukta özel emzikler kullanılır ya da
nazogastrik sonda konur. İri dil için erken yaşta yapılan dil küçültme
cerrahisi yararlı olur. İri dil nedeniyle konuşma zorluğu çeken çocuğa
konuşma terapisi uygulanır.
Yüzde asimetrik
büyüme görülen olguda kranyofasyal cerrahi teknikleri uygulanır.
8 ila 15 yaş arası
dönemde yılda iki kez idrarda kalsiyum/kreatinin oranına bakılır ve yılda
bir yapılacak böbrek ultrasonuyla nefrokalsinoz ve medüller sünger böbrek
varlığı araştırılır.
En az sekiz yaşına
kadar her üç ayda bir yapılacak batın ultrasonografisiyle Wilms tümörü
gelişip gelişmediği izlenir. Benzer şekilde en az dört yaşına kadar her üç
ayda bir serum alfa-fetoprotein seviyesine bakılarak hepatoblastom
gelişmediğinden emin olunur. Embriyoner tümör mevcutsa standart pediatrik
onkoloji protokollerine uyulur.
Ortopedik Tedavi. Alt
ekstremitede ciddi uzunluk farkı mevcutsa
tercihan ergenlik döneminde cerrahi olarak denkleştirme yöntemlerinden biri
tercih edilir (epifizyodez ya da
kallotazis).
Bu sayfada yer alan bilgilerin tamamı ebeveynleri çocuk ortopedisinin konuları hakkında bilgilendirmek amacıyla verilmektedir.
Bu bilgilerden yola çıkarak ebeveynlerin çocuklarındaki rahatsızlıklara tanı koymaları, daha da ileri giderek kendilerini hekim yerine koyarak çocuklarını tedavi etmeye kalkışmaları son derece sakıncalıdır.
Bu sayfada yer alan bilgiler bir hekimin muayene sonucu vereceği kararın yerini asla alamaz.