Klinik Görünüm
Marfan sendromuna ait belirti ve bulgular kişiden kişiye değişir. Çocukların
bazılarında klinik bulgular hafifken, ya da belirti ve bulgulardan sadece
birkaçı mevcutken, diğerlerinde sendromun birçok belirti ve bulgusu bulunur
ve hastalık şiddetli seyreder.
Olguların çoğunda klinik tablo çocuk yaş aldıkça oturur, ancak
azımsanmayacak kadar az bir kısmında ise Marfan sendromu daha çocuk süt
çocukluğu dönemindeyken bellidir ve hızla birçok organı etkiler.
Marfan sendromlu çocuklarda görünüm çok çapıcıdır: oldukça uzun boylu
çocukların aşırı uzun ekstremiteleri mevcuttur (dolikostenomeli). Kulaç
uzunluğu hastanın boyundan daha uzundur. Hasta bacak bacak üstüne attığında
ayağı bacağın üzerinden yere değer (bacak bacak üstü belirtisi). İnce ve
uzun ekstremitelerde distal kemiklerin aşırı uzaması orantısız bir görünüm
yaratır.
Hastalığa yakalanmamış aile bireyleri arasında uzun boylu olmaları dikkat
çeker. Erişkin yaşa gelindiğinde boy genellikle 1.90 m’yi aşar. Üst gövde
uzunluğu (simfiz
pubisten baş üstüne kadar olan mesafe) ile alt gövde
uzunluğu (simfiz pubisten ayak tabanına kadar olan mesafe) arasındaki oran
normalde 0.93’tür. Marfan sendromlu hastalarda bu oran çok azalır.
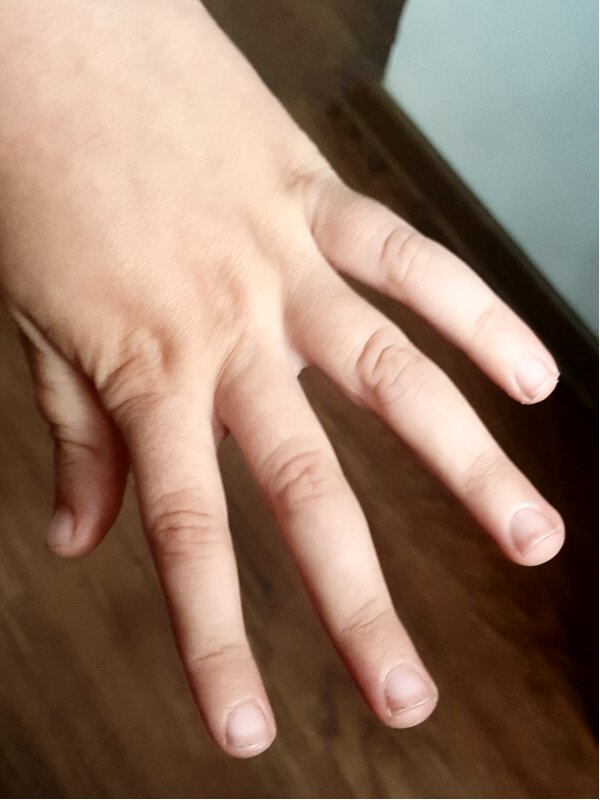
Marfan sendromlu çocukların el ve ayak parmakları örümceği andırır tarzda
ince ve aşırı uzundur (araknodaktili). Araknodaktili tanısı koyabilmek için
bazı testler tanımlanmıştır. Steinberg testinde başparmak avuç içine alınıp
yumruk yapıldığında başparmağın ucu 5. parmağın (küçük parmak) medialinde (iç
tarafında) gözükür (başparmak belirtisi). Steinberg testi Marfan sendromunda
rutin tarama testi olarak kabul edilmiştir. Ayrıca, Marfan sendromlu hasta
el bileğini diğer eliyle kavradığında başparmak 5. parmağın üzerine
biniyorsa Walker-Murdoch belirtisi müspet demektir (el-el bileği belirtisi).
Ekstremiteler hipotoniktir ve cilt altı yağ dokusu azdır. Cilt esnektir.
Ciltte kilo alıp vermeyle ilgisi olmayan çatlaklar (strialar) görülebilir.
Marfan sendromunda görülen çok sayıdaki iskelet malformasyonlarının başında
pektus ekskavatum (kunduracı göğsü) gelir. Kaburgaların aşırı uzamasıyla
sternum göğüs kafesinde gömülü kalırken göğüs kafesinin ön arka çapı azalır,
ya da bazen tersi gelişerek pektus karinatum (kuş göğsü) meydana gelir.
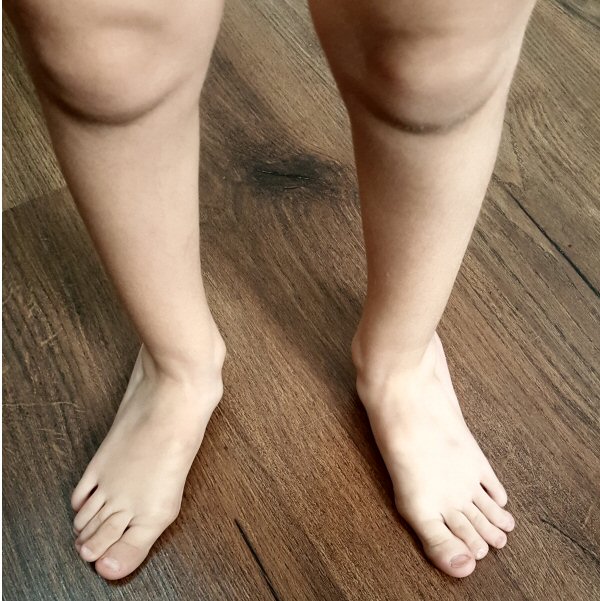
Bağ dokusunun esnek oluşu eklemlerde gevşekliğe (eklem laksitesi), belirgin
genu rekurvatuma (dizin geriye gitmesi) ve ileri derecede düz tabanlığa yol
açar. Ayak bağlarındaki esneklik sayesinde çocukluk çağında muayene
sırasında ayağı elle düzeltmek mümkün olur. Bazı hastalarda eklemler normal
olabilir; hatta kontraktüre yol açacak kadar sert bile olabilir (örneğin,
dirseği tam ekstansiyona getirmek mümkün olmayabilir). Bağ gevşekliği nadir
de olsa kalça çıkığına yol açabilir. El bileği bağlarının gevşekliği el
bileğinde çıkığa (perilunat çıkık) neden olabilir. Benzer mekanizmayla
patella çıkığı da görülebilir.
El parmaklarında kamptodaktili (genellikle küçük
parmakta görülen, parmağın proksimal interfalangeal ekleminde doğuştan
fleksiyon kontraktürü), ya da klinodaktili (genellikle küçük parmakta
görülen, doğuştan parmağın distal interfalangeal eklemden yandaki parmağa
doğru bükük oluşu) görülebilir.
Marfan sendromlu hastada femur başı derinleşen
asetabulumla birlikte giderek
pelvis boşluğuna gömülür (asetabuler protrüzyo). Olguların yaklaşık 4’te
1’inde görülen astabuler protrüzyo geç çocukluk ve ergenlik çağında ortaya
çıkar. Çoğu olguda ağrı bulunmaz, ancak kalça hareket açıklığı giderek az da
olsa azalır. Bazen femur başında gelişen erozyonlar
dejeneratif artrite
neden olur.
Hastaların çoğunda omurgaya ait sorun görülür. Bunların başında ilerleyici
skolyoz gelir (olguların yaklaşık % 30 ila % 100’ünde). Eğrilik çift eğrilik
tarzındadır; bazen daha fazla sayıda da olabilir. Skolyoza bağlı ağrı nadir
değildir. Bağ gevşekliğine bağlı
servikal
kifoz ve özellikle L5-S1
segmentinde olmak üzere spondilolistezis (bel kayması) görülebilir.
Bağ gevşekliği kasıkta, uylukta, diyafragmda ve ameliyat kesisinde fıtığa
neden olabilir.
Marfan sendromlu hastalarda uzun ve dar bir yüze yol açan kafatası yapısı (dolikosefali),
derine kaçan gözler (enoftalmi), oldukça ufak (mikrognati) ve üst çeneye
göre daha geride olan (retrognati) alt çene, elmacık kemiğinin hipoplazisine
bağlı düz yanaklar ve aşağı süzülen göz yapısından ibaret tipik bir yüz
görünümü vardır. Kafatasının yüksek oluşu frontal sinusun büyümesinin
sonucudur. Ayrıca, yüksek damak da görülebilir. Dişler bir araya
toplandığından hasta ısırdığında alt ve üst dişler aynı hizada kavuşmaz (maloklüzyon).
Marfan sendromlu hastada göz küresinin şeklindeki değişiklikler ileri
derecede miyopiye neden olur. Miyopi küçük yaşta ortaya çıkar ve giderek
artar. Hastaların yaklaşık % 60’ında gevşek, hatta kopuk asıcı mercek
bağları göz merceğinin (lens) disloke olmasına yol açar (ectopia lentis).
Çıkık tipik olarak yukarı ve dışa doğrudur. Lens çıkığı en iyi pupilla
dilatasyonu (gözbebeğinin genişlemesi) sonrasında dar ışık muayenesi ile
saptanır. Korneanın (saydam tabaka) anormal düzleşmesi, renkli iris
tabakasının hipoplazisi ve retina dekolmanı (ağtabakanın ayrılması) Marfan
sendromunda görülebilen diğer göz komplikasyonları arasında sayılabilir.
Bazı hastalarda erkenden katarakt, ya da
glokom gelişir.
Marfan sendromlu hastada yaşamı tehdit eden, ciddi kardiyovasküler
komplikasyonlar gelişebilir. Bunlar arasında en yaygın olanları
mitral kapak
prolapsusu ve çıkan aort dilatasyonudur.
Mitral kapak prolapsusu genelde bir belirtiye yol açmaz, ancak sol kalp
karıncığının kasılması sırasında mitral kapak yetersizliği nedeniyle sol
kulakçığa geri kaçan kan miktarı arttıkça göğüs ağrısı ve anormal kalp
ritmine (aritmi) neden olur. Zamanla konjestif kalp yetmezliği gelişir.
Damar yapısındaki gevşeklik sonucu oluşan çıkan aort
dilatasyonu (genişlemesi) aort kapağı yetmezliğine neden olur; yani, aorta
pompalanan kanın bir kısmı sol kulakçığa geri döner ve zamanla konjestif
kalp yetmezliği gelişir. Giderek dejenere olan genişlemiş aortta zamanla
damar duvarının iç ve dış tabakaları birbirinden ayrılır ve giderek dissekan
aort anevrizması gelişir.
Damar çapının normalden %50 daha fazla genişlemesine
yol açan bir cins balonlaşma olarak tanımlayabileceğimiz anevrizmanın
yırtılması Marfan sendromlu hastada en sık rastlanan ölüm sebebidir.
Dissekan
aort anevrizması aortun diğer kısımlarında da olabilir. Bazen akciğer
damarlarında da (pulmoner arterler) genişlemeler görülür.
Ani göğüs ağrısı ile ortaya çıkan spontan pnömotoraks (akciğerler ile göğüs
kafesi arasına hava toplanması) Marfan sendromlu hastalarda rastlanabilen
ciddi bir akciğer komplikasyonudur. Marfan sendromlu hastada akciğerlerin
apikal (tepe) kısmında oluşan hava boşluklarının plevra (akciğerleri saran
zar) boşluğuna
açılmasıyla oluşur. Pnömotoraks bazen tekrarlayabilir.
Marfan sendromlu hastada omuriliği saran dura kesesinde genişlemeler ve
omurilikten çıkan sinir köklerine doğu fıtıklaşmalar bulunur (dural ektazi).
Bu durum hemen her zaman
lomber bölgede olur. Hastaların yaklaşık % 60 ila %
90’ında rastlanan bu durum genellikle
asemptomatiktir, ancak bu durum bazen
bel ağrısına, bacaklara vuran kök ağrılarına, bacaklarda duyu bozukluklarına
(parestezi) ve kas güçsüzlüğüne neden olur.
Bu sayfada yer alan bilgilerin tamamı ebeveynleri çocuk ortopedisinin konuları hakkında bilgilendirmek amacıyla verilmektedir.
Bu bilgilerden yola çıkarak ebeveynlerin çocuklarındaki rahatsızlıklara tanı koymaları, daha da ileri giderek kendilerini hekim yerine koyarak çocuklarını tedavi etmeye kalkışmaları son derece sakıncalıdır.
Bu sayfada yer alan bilgiler bir hekimin muayene sonucu vereceği kararın yerini asla alamaz.